


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 -thrombin-bivalirubin complex at pD 5.0; Protonation states and hydration structure of the enzyme-product complex
-thrombin-bivalirubin complex at pD 5.0; Protonation states and hydration structure of the enzyme-product complexYamada, Taro*; Kurihara, Kazuo; Onishi, Yuki*; Tamada, Taro; Tomoyori, Katsuaki; Masumi, Kenji*; Tanaka, Ichiro*; Kuroki, Ryota; Niimura, Nobuo*
Biochimica et Biophysica Acta; Proteins and Proteomics, 1834(8), p.1532 - 1538, 2013/08
Times Cited Count:17 Percentile:48.69(Biochemistry & Molecular Biology)The protonation states and hydration structures of the -thrombin-bivalirubin complex were studied by joint XN refinement of the single crystal X-ray and neutron diffraction data at resolutions of 1.6 and 2.8 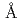 , respectively. The atomic distances were estimated by carrying out X-ray crystallographic analysis at 1.25 resolution. The complex represents a model of the enzyme-product (EP) complex of -thrombin. The neutron scattering length maps around the active site suggest that the side chain of H57/H was deuterated. The joint XN refinement showed that occupancies for D
, respectively. The atomic distances were estimated by carrying out X-ray crystallographic analysis at 1.25 resolution. The complex represents a model of the enzyme-product (EP) complex of -thrombin. The neutron scattering length maps around the active site suggest that the side chain of H57/H was deuterated. The joint XN refinement showed that occupancies for D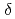 1 and D
1 and D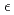 2 of H57/H were 1.0 and 0.7, respectively. However, no significant neutron scattering length density was observed around the hydroxyl oxygen O
2 of H57/H were 1.0 and 0.7, respectively. However, no significant neutron scattering length density was observed around the hydroxyl oxygen O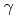 of S195/H, which was close to the carboxylic carbon atom of dFPR-COOH. These observations suggest that the O atom of S195/H is deprotonated and maintains its nucleophilicity in the EP complex. In addition to the active site, the hydration structures of the S1 subsite and the Exosite I, which are involved in the recognition of bivalirudin, are presented.
of S195/H, which was close to the carboxylic carbon atom of dFPR-COOH. These observations suggest that the O atom of S195/H is deprotonated and maintains its nucleophilicity in the EP complex. In addition to the active site, the hydration structures of the S1 subsite and the Exosite I, which are involved in the recognition of bivalirudin, are presented.
 protease I at pD 8.0; Protonation states and hydration structure in the free-form
protease I at pD 8.0; Protonation states and hydration structure in the free-formOnishi, Yuki*; Yamada, Taro*; Kurihara, Kazuo; Tanaka, Ichiro*; Sakiyama, Fumio*; Masaki, Takeharu*; Niimura, Nobuo*
Biochimica et Biophysica Acta; Proteins and Proteomics, 1834(8), p.1642 - 1647, 2013/08
Times Cited Count:8 Percentile:20.86(Biochemistry & Molecular Biology)The structure of the free-form of protease I (API) at pD 8.0 was refined by simultaneous use of single crystal X-ray and neutron diffraction data sets to investigate the protonation states of key catalytic residues of the serine protease. Occupancy refinement of the catalytic triad in the active site of API free-form showed that ca. 30% of the imidazole ring of H57 and ca. 70% of the hydroxyl group of S194 were deuterated. This observation indicates that a major fraction of S194 is protonated in the absence of a substrate. The protonation state of the catalytic triad in API was compared with the bovine 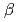 -trypsin-BPTI complex. The comparison led to the hypothesis that close contact of a substrate with S194 could lower the acidity of its hydroxyl group, thereby allowing H57 to extract the hydrogen from the hydroxyl group of S194. H210, which is a residue specific to API, does not form a hydrogen bond with the catalytic triad residue D113. Instead, H210 forms a hydrogen bond network with S176, H177 and a water molecule. The close proximity of the bulky, hydrophobic residue W169 may protect this hydrogen bond network, and this protection may stabilize the function of API over a wide pH range.
-trypsin-BPTI complex. The comparison led to the hypothesis that close contact of a substrate with S194 could lower the acidity of its hydroxyl group, thereby allowing H57 to extract the hydrogen from the hydroxyl group of S194. H210, which is a residue specific to API, does not form a hydrogen bond with the catalytic triad residue D113. Instead, H210 forms a hydrogen bond network with S176, H177 and a water molecule. The close proximity of the bulky, hydrophobic residue W169 may protect this hydrogen bond network, and this protection may stabilize the function of API over a wide pH range.
Nakagawa, Hiroshi; Kamikubo, Hironari*; Kataoka, Mikio
Biochimica et Biophysica Acta; Proteins and Proteomics, 1804(1), p.27 - 33, 2010/01
Times Cited Count:19 Percentile:47.02(Biochemistry & Molecular Biology)In order to examine the properties specific to the folded protein, the effect of the conformational states on protein dynamical transition was studied by incoherent elastic neutron scattering for both wild type and a deletion mutant of staphylococcal nuclease. The deletion mutant of SNase which lacks C-terminal 13 residues takes a compact denatured structure, and can regard as a model of intrinsic unstructured protein. Incoherent elastic neutron scattering experiments were carried out at various temperature between 10K and 300K on IN10 and IN13 installed at ILL. Temperature dependence of mean square displacements was obtained by the q-dependence of elastic scattering intensity. The measurements were performed on dried and hydrated powder samples. No significant differences were observed between wild type and the mutant for the hydrated samples, while significant differences were observed for the dried samples. A dynamical transition at 140K observed for both dried and hydrated samples. The slopes of the temperature dependence of MSD before transition and after transition are different between wild type and the mutant, indicating the folding induces hardening. The hydration water activates a further transition at 240K. The behavior of the temperature dependence of MSD is indistinguishable for wild type and the mutant, indicating that hydration water dynamics dominate the dynamical properties.
 PprA protein by Atomic force microscopy
PprA protein by Atomic force microscopyMurakami, Masahiro*; Narumi, Issei; Sato, Katsuya*; Furukawa, Akira*; Hayata, Isamu*
Biochimica et Biophysica Acta; Proteins and Proteomics, 1764(1), p.20 - 23, 2006/01
Times Cited Count:11 Percentile:22.1(Biochemistry & Molecular Biology)A DNA repair-promoting protein, PprA, was isolated from a radiation resistant bacterium, . Despite several studies, however, the function of PprA is not still clear. We used atomic force microscopy (AFM) to elucidate the role of this protein in the DNA repair pathway. In the present study, interaction between the linear DNA and PprA protein was imaged and analyzed by AFM without any fixation or staining. Though both end-bound and internally bound PprA was observed, the affinity of the end-bound protein was greater considering the proportion of features of binding analyzed by AFM. In some conditions, looping forms of the DNA-PprA complex were observed. Gel filtration high performance liquid chromatography (HPLC) was also conducted to estimate the molecular weight of this protein. The result of the HPLC analysis suggested that PprA formed multimers in buffer solution without DNA.
